Library Services
For labs that wish to perform their own library preparation,
Discovery Genomics will be happy to sequence prepared libraries. Individual indexed
libraries may be submitted for QC and pooling, or the libraries may be
pooled by the customer and sent as ready to run.
The following policies apply for user prepared libraries:
- User prepared libraries will first be subjected to the same three
QC assays that are performed on Discovery-prepared
libraries:
- Initial quantification via Qubit
- Bioanalysis using the Agilent 2100 Bioanalyzer or Caliper LabChip
GX - allows determination of the average library fragment size
and test for the presence of multiple peaks. Multiple peaks are a
strong indication of poor sequencing performance.
- Final quantification with a
Kapa Library Quant Kit - using a 10nM dilution (based on
Bioanalysis data), which independently validates concentration and is
used to determine final concentration for sequencing.
- Once the QC process has been completed, all QC-related data will be
posted to the project page. If all QC data indicates the library
should perform well, it will go into the sequencing queue.
- If the QC data shows anything unusual, Discovery will
contact the submitting investigator, who will then make the final
decision on whether to move forward with sequencing and the
appropriate sequencing conditions.
Discovery makes every effort to accurately determine library
concentration and the appropriate sequencing dilution using the above
protocol. However, for libraries created by other labs, there can be
difficulty in predicting the appropriate sequencing dilution, especially
if the libraries have multiple peaks or consist of pooled samples.
Discovery will document the quantitation process for the
libraries in question and make the best decision given the available
information. This may include spiking in PhiX to increase base
diversity, clustering the library at a lower than standard density, or
any other sequencer manipulation that may increase the chances of a
successful run (see Special Library Considerations). However, the final
decision on whether a library is sequenced (and under what conditions)
will rest with the customer and Discovery Genomics is not responsible
for failed sequencing if it is determined to be due to the library
rather than a sequencer problem.
Discovery Genomics cannot
guarantee performance or sequencing output for libraries generated by
customers; therefore payment is required prior to sequencing.
Please read our
polices
for more information.
Special Library Considerations
- Libraries cannot be indexed (barcoded) after preparation, it must be done as part of the library prep protocol. Therefore please contact us if you are interested in indexing your samples.
- Discovery supports several forms of indexing, including Illumina Tru-Seq, Nextera, NEB, and BIOO. Some protocols may require custom sequencing primers and it is your responsibility to provide the primers (at 100µM) and instructions for use (i.e. custom read 1 primer, or indexing primer, etc) at the time of submission.
- Not all forms of indexing or library preparation are appropriate for current sequencer software versions. The user is responsible for disclosing all relevant library prep details before sequencing is attempted to avoid disappointment in data output. For example, certain library construction protocols can result in identical sequences for the first several bases of each read, which causes problems for the software when it tries to define the cluster locations. Previous versions of the Illumina software could defer basecalling for a given number of cycles to avoid this problem, but this capability no longer exists. For such libraries, the only options for accurately sequencing such libraries are:
- Mix with another library from the same user and demultiplex reads after sequencing
- Mix with Phix and lose a fraction of the usable reads (normally 25%)
- Sequence at a low cluster density so that the clusters are spatially separate from each other
- Choose a sequencing platform that handles low diversity well. For example, the MiSeq handles amplicon sequencing very well.
- User-prepared libraries are run on the lane or flow cell level.
That is, outside libraries are not mixed with Discovery-prepared
libraries to share a lane. If multiple types of libraries are
pooled together (for example, ChIP-seq and RNA-seq) the output will
most likely not be as balanced as attended. Library size,
preparation method, and library quality all affect the clustering
stage of sequencing and those factors cannot be accounted for by QC
measures.
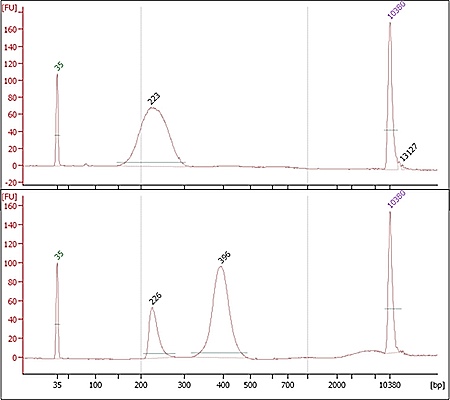
Electropherograms for two libraries prepared by Discovery clients. The top library has the expected profile, with a single peak of fragment sizes. The bottom library is likely overamplified, with a secondary peak around twice the size of the expected fragments. Occasionally dimer will appear as smaller peaks close to 120bp. If adapter dimer is seen in a library, Discovery staff will evaluate how much is present and whether it could affect the clustering or sequencing quality of the library. We may recommend a bead clean-up be performed for an additional fee to increase the chances of sequencing success.
Sample Submission Requirements
Libraries should be submitted at 15nM concentration in 15mM Tris (pH 8.0), and at least 15µl should be provided for ease of handling. Libraries submitted in less than 15ul of volume will be
subject to an additional handling fee. You may send your final library without attempting to dilute it to 15nM as long as you are submitting at least 15ul, since it is always easier for us to dilute a library than to concentrate it without the risk of compromising its integrity. The lowest concentration of library appropriate for standard clustering SOPs is 1.25nM. We do have custom protocols available for sequencing even lower concentration libraries, but they are considered experimental and run at the customer's risk. Please discuss these projects with a Discovery staff member before submitting. When libraries are submitted, please provide as much information about their construction as possible, as it helps us provide you with the data you expect. If your libraries require a custom sequencing or index primer, you must provide this with your libraries at 100uM concentration.
Library Data
Data output for library submission is demultiplexed fastq files. Additional analysis, such as alignment to a standard genome, can also be requested for an additional fee.
