All nucleic acids received by Discovery Genomics are subject to a QC/QA procedure that is designed to
identify possible low-performing samples before they are used in protocols. If samples do
not meet minimum standards, replacement samples can be submitted with new sample IDs. Discovery Genomics
will not pool samples for any reason.
What happens if your samples do not meet our requirements for
input amounts and/or concentration?
You will receive an email detailing the QC findings and requesting your
review of the data.
Your project will be put on hold until a permission to proceed is
received.
You may submit replacement samples with new Discovery numbers
through your project. Samples already submitted will be held to process
all samples as a batch.
You may choose to proceed with samples that have lower than required
amounts and/or subpar integrity. We require this confirmation in writing
with your acknowledgement that we cannot warranty the results.
What is the effect of low input or poor quality samples on the results?
Discovery Genomics can almost always make a library out of any amount of nucleic acid. The output of such libraries will normally have an expected bioanalysis profile and some nM concentration as measured by kapa qPCR as part of the final library QC. We always monitor metrics throughout the library preparation and flag any samples that are behaving aberrantly, but in most cases there will be no discernable warning that a library should not proceed to sequencing. If we do see low yield, unusual profiles, or other problems, those libraries will be discussed with you as potential libraries to be pulled from sequencing.
Most effects of low input or degraded sample will only be seen in the final sequencing data. We can usually generate the amount of sequencing requested for a sub-par sample, but cannot know how complex the library that we created will be. The complexity of the library and the duplication rate will directly impact the coverage generated for both DNA and RNA-based projects.
We understand that many nucleic acids isolated from FFPE samples will be poor quality, both degraded (short fragments instead of intact DNA) and possibly low input. We have modified protocols designed to maximize the chances of success when working with such samples; however, we cannot guarantee performance of library preparation or final data quality and complexity for nucleic acids that do not pass our initial QC. We are pleased to offer processing of subpar samples as a service as many FFPE samples are both valuable and irreplacable, but the customer must assume the risks involved. Payment for services rendered is expected regardless of outcome when working with samples that do not meet our QC specifications.
If you know your samples will likely be degraded or low input, inform us of that at the time of submission in the Special Instructions and request modified protocols, including conservation of material used during QC. You will still be contacted at the completion of QC to review the results and grant us permission to proceed.
FFPE DNA samples tend to generate better exome data than WGS data, as the capture process reduces the complexity of the library. For both WGS and exome, degradation is less of a worry than low input. The more input you can provide for FFPE samples, the better your results should be.
FFPE RNA samples must usually be processed by our ribosomal reduction RNA-seq protocol as they are usually too degraded for a polyA selection to be successful. Amplification is not recommended as its success relies on intact RNA. Small RNA-seq protocols are not likely to be specific as the size of the degraded fragments may co-localize during the gel extraction step.
DNA Quality Control
DNA QC includes:
Quantification using PicoGreen® or Qubit® fluorescence based assays
Visualization of DNA size on an agarose gel to check for degradation
Nanodrop concentration determination routinely over-estimates the amount of DNA in a
sample since it is not dsDNA-specific and can be thrown off by contaminants. Therefore,
if using nanodrop for quantification of your samples, be sure to aim for the top range
of material requested as it is likely your total amounts will be 30-50% lower than expected.
RNA Quality Control
RNA QC includes:
Quantification via Qubit®
Bioanalysis using the Agilent 2100 Bioanalyzer
In bioanalysis, the electropherogram of a total RNA sample is analyzed and assigned an
RNA Integrity Number, or RIN. The highest quality RNA will have RIN scores of 10, with
samples having RINs of 7.0 or greater generally performing well in most protocols. Non-mammalian
species are often assigned lower RIN numbers due to variation in expected ribosomal RNA sizes.
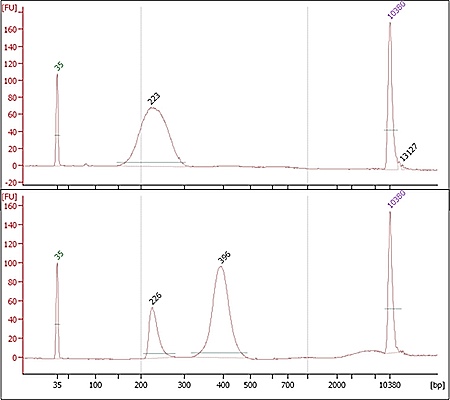
Agilent 2100 electropherograms for two high-quality RNA samples. The top image is human and
received a RIN of 10.0, while the bottom sample is non-mammalian, and the unexpected size of
rRNA molecules caused scoring to fail.
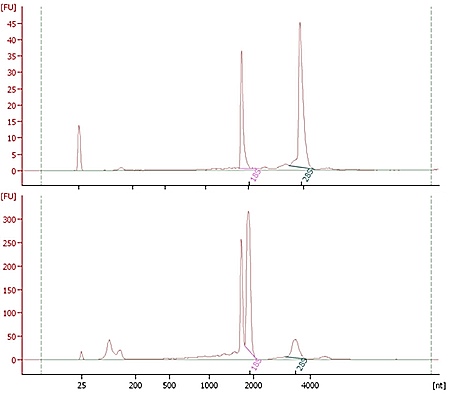
Electropherograms for two libraries prepared by Discovery clients. The top library has the expected
profile, with a single peak of fragment sizes. The bottom library is likely overamplified,
with a secondary peak around twice the size of the expected fragments.
Bioanalysis using the Agilent 2100 Bioanalyzer to check library peak size and for
the presence of multiple peaks
Kapa RT-PCR on a 10nM dilution to independently validate concentration and determine
final concentration for sequencing
Using the above protocol, Discovery Genomics makes every effort to accurately determine library
concentration and the appropriate sequencing dilution. However, for libraries created by
other labs, there is sometimes difficulty in predicting the appropriate dilution, especially
if the libraries have multiple peaks or consist of pooled samples. Discovery Genomics will document the
quantitation process for the libraries in question and the final decision on sequencing
concentration will rest with the user.
Discovery Genomics cannot guarantee performance or sequencing output for libraries generated by outside users
; therefore payment is required prior to sequencing.